Pfizer Inc.
Attention: Ms. Elisa Harkins
500 Arcola Road
Collegeville, PA 19426
Dear Ms. Harkins:
On February 4, 2020, pursuant to Section
564(b)(1)(C) of the Federal Food, Drug, and
Cosmetic Act (the FD&C Act or the Act),
the Secretary of the Department of Health and
Human Services (HHS) determined that there is
a public health emergency that has a
significant potential to affect national
security or the health and security of United
States citizens living abroad, and that
involves the virus that causes Coronavirus
Disease 2019 (COVID-19).1 On the basis of such
determination, the Secretary of HHS on March
27, 2020, declared that circumstances exist
justifying the authorization of emergency use
of drugs and biological products during the
COVID-19 pandemic, pursuant to Section 564 of
the Act (21 U.S.C. 360bbb-3), subject to terms
of any authorization issued under that
section.2
On December 11, 2020, the Food and Drug
Administration (FDA) issued an Emergency Use
Authorization (EUA) for emergency use of
Pfizer-BioNTech COVID‑19 Vaccine for the
prevention of COVID-19 for individuals 16
years of age and older pursuant to Section 564
of the Act. FDA reissued the letter of
authorization on: December 23, 2020,3 February
25, 2021,4 May
1 U.S. Department of Health and Human
Services, Determination of a Public Health
Emergency and Declaration that Circumstances
Exist Justifying Authorizations Pursuant to
Section 564(b) of the Federal Food, Drug, and
Cosmetic Act, 21 U.S.C. § 360bbb-3. February
4, 2020.
2 U.S. Department of Health and Human
Services, Declaration that Circumstances Exist
Justifying Authorizations Pursuant to Section
564(b) of the Federal Food, Drug, and Cosmetic
Act, 21 U.S.C. § 360bbb-3, 85 FR 18250 (April
1, 2020).
3 In the December 23, 2020 revision, FDA
removed reference to the number of doses per
vial after dilution from the letter of
authorization, clarified the instructions for
vaccination providers reporting to VAERS, and
made other technical corrections. FDA also
revised the Fact Sheet for Healthcare
Providers Administering Vaccine (Vaccination
Providers) to clarify the number of doses of
vaccine per vial after dilution and the
instructions for reporting to VAERS. In
addition, the Fact Sheet for Healthcare
Providers Administering Vaccine (Vaccination
Providers) and the Fact Sheet for Recipients
and Caregivers were revised to include
additional information on safety monitoring
and to clarify information about the
availability of other COVID-19 vaccines.
4 In the February 25, 2021 revision, FDA
allowed flexibility on the date of submission
of monthly periodic safety reports and revised
the requirements for reporting of vaccine
administration errors by Pfizer Inc. The Fact
Sheet for Health Care Providers Administering
Vaccine (Vaccination Providers) was revised to
provide an update to the storage and
transportation temperature for frozen vials,
direct the provider to the correct CDC website
for information on monitoring vaccine
recipients for the occurrence of immediate
adverse reactions, to include data from a
developmental toxicity study, and add adverse
reactions that have been identified during
post authorization use. The Fact Sheet for
Recipients and Caregivers was revised to add
adverse reactions that have been identified
during post authorization use.
Page 2 – Pfizer Inc.
10, 2021,
5 June 25, 2021,6 and August 12, 2021.7
On August 23, 2021, FDA approved the biologics
license application (BLA) submitted by
BioNTech Manufacturing GmbH for COMIRNATY
(COVID-19 Vaccine, mRNA) for active
immunization to prevent COVID-19 caused by
SARS-CoV-2 in individuals 16 years of age and
older.
On August 23, 2021, having concluded that
revising this EUA is appropriate to protect
the public health or safety under section
564(g)(2) of the Act, FDA is reissuing the
August 12, 2021 letter of authorization in its
entirety with revisions incorporated to
clarify that the EUA will remain in place for
the Pfizer-BioNTech COVID-19 vaccine for the
previously-authorized indication and uses, and
to authorize use of COMIRNATY (COVID-19
Vaccine, mRNA) under this EUA for certain uses
that are not included in the approved BLA. In
addition, the Fact Sheet for Healthcare
Providers Administering Vaccine (Vaccination
Providers) was revised to provide updates on
expiration dating of the authorized
Pfizer-BioNTech COVID-19 Vaccine and to update
language regarding warnings and precautions
related to myocarditis and pericarditis. The
Fact Sheet for Recipients and Caregivers was
updated as the Vaccine Information Fact Sheet
for Recipients and Caregivers, which comprises
the Fact Sheet for the authorized
Pfizer-BioNTech COVID-19 Vaccine and
information about the FDA-licensed vaccine,
COMIRNATY (COVID-19 Vaccine, mRNA).
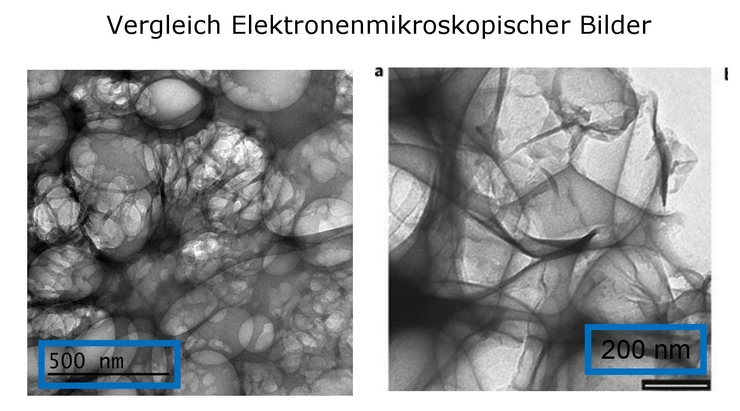
Pfizer-BioNTech COVID‑19 Vaccine contains a
nucleoside-modified messenger RNA (modRNA)
encoding the viral spike (S) glycoprotein of
SARS-CoV-2 formulated in lipid particles.
COMIRNATY (COVID-19 Vaccine, mRNA) is the same
formulation as the Pfizer-BioNTech COVID-19
Vaccine and can be used interchangeably with
the Pfizer-BioNTech COVID-19 Vaccine to
provide the COVID-19 vaccination series.8
5 In the May 10, 2021 revision, FDA authorized
Pfizer-BioNTech Vaccine for the prevention of
COVID-19 in individuals 12 through 15 years of
age, as well as for individuals 16 years of
age and older. In addition, FDA revised the
Fact Sheet for Healthcare Providers
Administering Vaccine (Vaccination Providers)
to include the following Warning: “Syncope
(fainting) may occur in association with
administration of injectable vaccines, in
particular in adolescents. Procedures should
be in place to avoid injury from fainting.” In
addition, the Fact Sheet for Recipients and
Caregivers was revised to instruct vaccine
recipients or their caregivers to tell the
vaccination provider about fainting in
association with a previous injection.
6 In the June 25, 2021 revision, FDA clarified
terms and conditions that relate to export of
Pfizer-BioNTech COVID‑19 Vaccine from the
United States. In addition, the Fact Sheet for
Healthcare Providers Administering Vaccine
(Vaccination Providers) was revised to include
a Warning about myocarditis and pericarditis
following administration of the
Pfizer-BioNTech COVID-19 Vaccine. The Fact
Sheet for Recipients and Caregivers was
updated to include information about
myocarditis and pericarditis following
administration of the Pfizer-BioNTech COVID‑19
Vaccine.
7 In the August 12, 2021 revision, FDA
authorized a third dose of the Pfizer-BioNTech
COVID-19 Vaccine administered at least 28 days
following the two dose regimen of this vaccine
in individuals 12 years of age or older who
have undergone solid organ transplantation, or
individuals 12 years of age or older who are
diagnosed with conditions that are considered
to have an equivalent level of
immunocompromise.
8 The licensed vaccine has the same
formulation as the EUA-authorized vaccine and
the products can be used interchangeably to
provide the vaccination series without
presenting any safety or effectiveness
concerns. The products are legally distinct
with certain differences that do not impact
safety or effectiveness.
Page 3 – Pfizer Inc.
For the December 11, 2020 authorization for
individuals 16 years of age and older, FDA
reviewed safety and efficacy data from an
ongoing phase 1/2/3 trial in approximately
44,000 participants randomized 1:1 to receive
Pfizer-BioNTech COVID‑19 Vaccine or saline
control.
The trial has enrolled participants 12 years
of age and older. FDA’s review at that time
considered the safety and effectiveness data
as they relate to the request for emergency
use authorization in individuals 16 years of
age and older.
FDA’s review of the available safety data from
37,586 of the participants 16 years of age and
older, who were followed for a median of two
months after receiving the second dose, did
not identify specific safety concerns that
would preclude issuance of an EUA. FDA’s
analysis of the available efficacy data from
36,523 participants 12 years of age and older
without evidence of SARS-CoV-2 infection prior
to 7 days after dose 2 confirmed the vaccine
was 95% effective (95% credible interval 90.3,
97.6) in preventing COVID-19 occurring at
least 7 days after the second dose (with 8
COVID-19 cases in the vaccine group compared
to 162 COVID-19 cases in the placebo group).
Based on these data, and review of
manufacturing information regarding product
quality and consistency, FDA concluded that it
is reasonable to believe that Pfizer-BioNTech
COVID‑19 Vaccine may be effective.
Additionally, FDA determined it is reasonable
to conclude, based on the totality of the
scientific evidence available, that the known
and potential benefits of Pfizer-BioNTech
COVID‑19 Vaccine outweigh the known and
potential risks of the vaccine, for the
prevention of COVID-19 in individuals 16 years
of age and older. Finally, on December 10,
2020, the Vaccines and Related Biological
Products Advisory Committee voted in agreement
with this conclusion.
For the May 10, 2021 authorization for
individuals 12 through 15 years of age, FDA
reviewed safety and effectiveness data from
the above-referenced, ongoing Phase 1/2/3
trial that has enrolled approximately 46,000
participants, including 2,260 participants 12
through 15 years of age. Trial participants
were randomized 1:1 to receive Pfizer-BioNTech
COVID-19 Vaccine or saline control. FDA’s
review of the available safety data from 2,260
participants 12 through 15 years of age, who
were followed for a median of 2 months after
receiving the second dose, did not identify
specific safety concerns that would preclude
issuance of an EUA. FDA’s analysis of
SARS-CoV-2 50% neutralizing antibody titers 1
month after the second dose of Pfizer-BioNTech
COVID-19 Vaccine in a subset of participants
who had no serological or virological evidence
of past SARS-CoV-2 infection confirm the
geometric mean antibody titer in participants
12 through 15 years of age was non-inferior to
the geometric mean antibody titer in
participants 16 through 25 years of age. FDA’s
analysis of available descriptive efficacy
data from 1,983 participants 12 through 15
years of age without evidence of SARS-CoV-2
infection prior to 7 days after dose 2 confirm
that the vaccine was 100% effective (95%
confidence interval 75.3, 100.0) in preventing
COVID-19 occurring at least 7 days after the
second dose (with no COVID-19 cases in the
vaccine group compared to 16 COVID-19 cases in
the placebo group). Based on these data, FDA
concluded that it is reasonable to believe
that Pfizer-BioNTech COVID‑19 Vaccine may be
effective in individuals 12 through 15 years
of age. Additionally, FDA determined it is
reasonable to conclude, based on the totality
of the scientific evidence available, that the
known and potential benefits of
Pfizer-BioNTech COVID‑19 Vaccine outweigh the
known and potential risks of the vaccine, for
the prevention of COVID-19 in individuals 12
through 15 years of age.
Page 4 – Pfizer Inc.
For the August 12, 2021 authorization of a
third dose of the Pfizer-BioNTech COVID-19
Vaccine in individuals 12 years of age or
older who have undergone solid organ
transplantation, or individuals 12 years of
age or older who are diagnosed with conditions
that are considered to have an equivalent
level of immunocompromise, FDA reviewed safety
and effectiveness data reported in two
manuscripts on solid organ transplant
recipients. The first study was a single arm
study conducted in 101 individuals who had
undergone various solid organ transplant
procedures (heart, kidney, liver, lung,
pancreas) a median of 97±8 months earlier. A
third dose of the Pfizer-BioNTech COVID-19
Vaccine was administered to 99 of these
individuals approximately 2 months after they
had received a second dose. Levels of total
SARS-CoV-2 binding antibodies meeting the
pre-specified criteria for success occurred
four weeks after the third dose in 26/59
(44.0%) of those who were initially considered
to be seronegative and received a third dose
of the Pfizer-BioNTech COVID-19 Vaccine; 67/99
(68%) of the entire group receiving a third
vaccination were subsequently considered to
have levels of antibodies indicative of a
significant response. In those who received a
third vaccine dose, the adverse event profile
was similar to that after the second dose and
no grade 3 or grade 4 events were reported. A
supportive secondary study describes a
double-blind, randomized-controlled study
conducted in 120 individuals who had undergone
various solid organ transplant procedures
(heart, kidney, kidney-pancreas, liver, lung,
pancreas) a median of 3.57 years earlier
(range 1.99-6.75 years). A third dose of a
similar mRNA vaccine (the Moderna COVID-19
vaccine) was administered to 60 individuals
approximately 2 months after they had received
a second dose (i.e., doses at 0, 1 and 3
months); saline placebo was given to 60
individuals or comparison. The primary outcome
was anti-RBD antibody at 4 months greater than
100 U/mL. This titer was selected based on NHP
challenge studies as well as a large clinical
cohort study to indicate this antibody titer
was protective. Secondary outcomes were based
on a virus neutralization assay and
polyfunctional T cell responses. Baseline
characteristics were comparable between the
two study arms as were pre-intervention
anti-RBD titer and neutralizing antibodies.
Levels of total SARS-CoV-2 binding antibodies
indicative of a significant response occurred
four weeks after the third dose in 33/60
(55.0%) of the Moderna COVID-19 vaccinated
group and 10/57 (17.5%) of the placebo
individuals. In the 60 individuals who
received a third vaccine dose, the adverse
event profile was similar to that after the
second dose and no grade 3 or grade 4 adverse
events were reported. Despite the moderate
enhancement in antibody titers, the totality
of data (i.e., supportive paper by Hall et al.
demonstrated efficacy of the product in the
elderly and persons with co-morbidities)
supports the conclusion that a third dose of
the Pfizer-BioNTech COVID-19 vaccine may be
effective in this population, and that the
known and potential benefits of a third dose
of Pfizer-BioNTech COVID-19 Vaccine outweigh
the known and potential risks of the vaccine
for immunocompromised individuals at least 12
years of age who have received two doses of
the Pfizer-BioNTech COVID-19 Vaccine and who
have undergone solid organ transplantation, or
who are diagnosed with conditions that are
considered to have an equivalent level of
immunocompromise.
Having concluded that the criteria for
issuance of this authorization under Section
564(c) of the Act are met, I am authorizing
the emergency use of Pfizer-BioNTech COVID‑19
Vaccine for the prevention of COVID-19, as
described in the Scope of Authorization
section of this letter (Section II) and
subject to the terms of this authorization.
Additionally, as specified in subsection
III.BB, I am authorizing use of COMIRNATY
(COVID-19 Vaccine, mRNA) under this EUA when
used to provide a two-dose regimen for
individuals aged 12 through 15 years, or
Page 5 – Pfizer Inc.
to provide a third dose to individuals 12
years of age or older who have undergone solid
organ transplantation or who are diagnosed
with conditions that are considered to have an
equivalent level of immunocompromise.
I. Criteria for Issuance of Authorization
I have concluded that the emergency use of
Pfizer-BioNTech COVID‑19 Vaccine for the
prevention of COVID-19 when administered as
described in the Scope of Authorization
(Section II) meets the criteria for issuance
of an authorization under Section 564(c) of
the Act, because:
A. SARS-CoV-2 can cause a serious or
life-threatening disease or condition,
including severe respiratory illness, to
humans infected by this virus;
B. Based on the totality of scientific
evidence available to FDA, it is reasonable to
believe that Pfizer-BioNTech COVID‑19 Vaccine
may be effective in preventing COVID-19, and
that, when used under the conditions described
in this authorization, the known and potential
benefits of Pfizer-BioNTech COVID‑19 Vaccine
when used to prevent COVID-19 outweigh its
known and potential risks; and
C. There is no adequate, approved, and
available9 alternative to the emergency use of
Pfizer-BioNTech COVID‑19 Vaccine to prevent
COVID-19.10
II. Scope of Authorization
I have concluded, pursuant to Section
564(d)(1) of the Act, that the scope of this
authorization is limited as follows:
• Pfizer Inc. will supply Pfizer-BioNTech
COVID‑19 Vaccine either directly or through
authorized distributor(s),11 to emergency
response stakeholders12 as directed by the
U.S.
9 Although COMIRNATY (COVID-19 Vaccine, mRNA)
is approved to prevent COVID-19 in individuals
16 years of age and older, there is not
sufficient approved vaccine available for
distribution to this population in its
entirety at the time of reissuance of this
EUA. Additionally, there are no products that
are approved to prevent COVID-19 in
individuals age 12 through 15, or that are
approved to provide an additional dose to the
immunocompromised population described in this
EUA.
10 No other criteria of issuance have been
prescribed by regulation under Section
564(c)(4) of the Act.
11 “Authorized Distributor(s)” are identified
by Pfizer Inc. or, if applicable, by a U.S.
government entity, such as the Centers for
Disease Control and Prevention (CDC) and/or
other designee, as an entity or entities
allowed to distribute authorized
Pfizer-BioNTech COVID‑19 Vaccine.
12 For purposes of this letter, “emergency
response stakeholder” refers to a public
health agency and its delegates that have
legal responsibility and authority for
responding to an incident, based on political
or geographical boundary lines (e.g., city,
county, tribal, territorial, State, or
Federal), or functional (e.g., law enforcement
or public health range) or sphere of authority
to administer, deliver, or distribute vaccine
in an emergency situation. In some cases
(e.g., depending on a state or local
jurisdiction’s COVID-19 vaccination response
organization and plans), there might be
overlapping roles and responsibilities among
“emergency response stakeholders” and
“vaccination providers” (e.g., if a local
health department is administering COVID-19
vaccines; if a pharmacy is acting in an
Page 6 – Pfizer Inc.
government, including the Centers for Disease
Control and Prevention (CDC) and/or other
designee, for use consistent with the terms
and conditions of this EUA;
• The Pfizer-BioNTech COVID‑19 Vaccine covered
by this authorization will be administered by
vaccination providers13 and used only to
prevent COVID-19 in individuals ages 12 and
older; and
• Pfizer-BioNTech COVID‑19 Vaccine may be
administered by a vaccination provider without
an individual prescription for each vaccine
recipient.
This authorization also covers the use of the
licensed COMIRNATY (COVID-19 Vaccine, mRNA)
product when used to provide a two-dose
regimen for individuals aged 12 through 15
years, or to provide a third dose to
individuals 12 years of age or older who have
undergone solid organ transplantation or who
are diagnosed with conditions that are
considered to have an equivalent level of
immunocompromise.
Product Description
The Pfizer-BioNTech COVID-19 Vaccine is
supplied as a frozen suspension in multiple
dose vials; each vial must be diluted with 1.8
mL of sterile 0.9% Sodium Chloride Injection,
USP prior to use to form the vaccine. The
Pfizer-BioNTech COVID-19 Vaccine does not
contain a preservative.
Each 0.3 mL dose of the Pfizer-BioNTech
COVID-19 Vaccine contains 30 mcg of a
nucleoside-modified messenger RNA (modRNA)
encoding the viral spike (S) glycoprotein of
SARS-CoV-2. Each dose of the Pfizer-BioNTech
COVID-19 Vaccine also includes the following
ingredients: lipids (0.43 mg
(4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate),
0.05 mg 2[(polyethylene
glycol)-2000]-N,N-ditetradecylacetamide, 0.09
mg 1,2-distearoyl-sn-glycero-3-phosphocholine,
and 0.2 mg cholesterol), 0.01 mg potassium
chloride, 0.01 mg monobasic potassium
phosphate, 0.36 mg sodium chloride, 0.07 mg
dibasic sodium phosphate dihydrate, and 6 mg
sucrose. The diluent (0.9% Sodium Chloride
Injection) contributes an additional 2.16 mg
sodium chloride per dose.
official capacity under the authority of the
state health department to administer COVID-19
vaccines). In such cases, it is expected that
the conditions of authorization that apply to
emergency response stakeholders and
vaccination providers will all be met.
13 For purposes of this letter, “vaccination
provider” refers to the facility,
organization, or healthcare provider licensed
or otherwise authorized by the emergency
response stakeholder (e.g., non-physician
healthcare professionals, such as nurses and
pharmacists pursuant to state law under a
standing order issued by the state health
officer) to administer or provide vaccination
services in accordance with the applicable
emergency response stakeholder’s official
COVID-19 vaccination and emergency response
plan(s) and who is enrolled in the CDC
COVID-19 Vaccination Program. If the vaccine
is exported from the United States, a
“vaccination provider” is a provider that is
authorized to administer this vaccine in
accordance with the laws of the country in
which it is administered. For purposes of this
letter, “healthcare provider” also refers to a
person authorized by the U.S. Department of
Health and Human Services (e.g., under the
PREP Act Declaration for Medical
Countermeasures against COVID-19) to
administer FDA-authorized COVID-19 vaccine
(e.g., qualified pharmacy technicians and
State-authorized pharmacy interns acting under
the supervision of a qualified pharmacist).
See, e.g., HHS. Fourth Amendment to the
Declaration Under the Public Readiness and
Emergency Preparedness Act for Medical
Countermeasures Against COVID-19 and
Republication of the Declaration. 85 FR 79190
(December 9, 2020).
Page 7 – Pfizer Inc.
The dosing regimen is two doses of 0.3 mL
each, 3 weeks apart. A third dose may be
administered at least 28 days following the
second dose of the two dose regimen of this
vaccine to individuals 12 years of age or
older who have undergone solid organ
transplantation, or individuals 12 years of
age or older who are diagnosed with conditions
that are considered to have an equivalent
level of immunocompromise.
The manufacture of the authorized
Pfizer-BioNTech COVID‑19 Vaccine is limited to
those facilities identified and agreed upon in
Pfizer’s request for authorization.
The Pfizer-BioNTech COVID-19 Vaccine vial
label and carton labels are clearly marked for
“Emergency Use Authorization.” The
Pfizer-BioNTech COVID‑19 Vaccine is authorized
to be distributed, stored, further
redistributed, and administered by emergency
response stakeholders when packaged in the
authorized manufacturer packaging (i.e., vials
and cartons), despite the fact that the vial
and carton labels may not contain information
that otherwise would be required under the
FD&C Act.
Pfizer-BioNTech COVID‑19 Vaccine is authorized
for emergency use with the following
product-specific information required to be
made available to vaccination providers and
recipients, respectively (referred to as
“authorized labeling”):
• Fact Sheet for Healthcare Providers
Administering Vaccine (Vaccination Providers):
Emergency Use Authorization (EUA) of
Pfizer-BioNTech COVID‑19 Vaccine to Prevent
Coronavirus Disease 2019 (COVID-19)
• Vaccine Information Fact Sheet for
Recipients and Caregivers About COMIRNATY
(COVID-19 Vaccine, mRNA) and Pfizer-BioNTech
COVID-19 Vaccine to Prevent Coronavirus
Disease (COVID-19).
I have concluded, pursuant to Section
564(d)(2) of the Act, that it is reasonable to
believe that the known and potential benefits
of Pfizer-BioNTech COVID‑19 Vaccine, when used
to prevent COVID-19 and used in accordance
with this Scope of Authorization (Section II),
outweigh its known and potential risks.
I have concluded, pursuant to Section
564(d)(3) of the Act, based on the totality of
scientific evidence available to FDA, that it
is reasonable to believe that Pfizer-BioNTech
COVID‑19 Vaccine may be effective in
preventing COVID-19 when used in accordance
with this Scope of Authorization (Section II),
pursuant to Section 564(c)(2)(A) of the Act.
Having reviewed the scientific information
available to FDA, including the information
supporting the conclusions described in
Section I above, I have concluded that
Pfizer-BioNTech COVID‑19 Vaccine (as described
in this Scope of Authorization (Section II))
meets the criteria set forth in Section 564(c)
of the Act concerning safety and potential
effectiveness.
The emergency use of Pfizer-BioNTech COVID‑19
Vaccine under this EUA must be consistent
with, and may not exceed, the terms of the
Authorization, including the Scope of
Authorization (Section II) and the Conditions
of Authorization (Section III). Subject to the
terms of this EUA and
Page 8 – Pfizer Inc.
under the circumstances set forth in the
Secretary of HHS’s determination under Section
564(b)(1)(C) described above and the Secretary
of HHS’s corresponding declaration under
Section 564(b)(1), Pfizer-BioNTech COVID‑19
Vaccine is authorized to prevent COVID-19 in
individuals 12 years of age and older as
described in the Scope of Authorization
(Section II) under this EUA, despite the fact
that it does not meet certain requirements
otherwise required by applicable federal law.
III. Conditions of Authorization
Pursuant to Section 564 of the Act, I am
establishing the following conditions on this
authorization:
Pfizer Inc. and Authorized Distributor(s)
A. Pfizer Inc. and authorized distributor(s)
will ensure that the authorized
Pfizer-BioNTech COVID‑19 Vaccine is
distributed, as directed by the U.S.
government, including CDC and/or other
designee, and the authorized labeling (i.e.,
Fact Sheets) will be made available to
vaccination providers, recipients, and
caregivers consistent with the terms of this
letter.
B. Pfizer Inc. and authorized distributor(s)
will ensure that appropriate storage and cold
chain is maintained until delivered to
emergency response stakeholders’ receipt
sites.
C. Pfizer Inc. will ensure that the terms of
this EUA are made available to all relevant
stakeholders (e.g., emergency response
stakeholders, authorized distributors, and
vaccination providers) involved in
distributing or receiving authorized
Pfizer-BioNTech COVID‑19 Vaccine. Pfizer Inc.
will provide to all relevant stakeholders a
copy of this letter of authorization and
communicate any subsequent amendments that
might be made to this letter of authorization
and its authorized labeling.
D. Pfizer Inc. may develop and disseminate
instructional and educational materials (e.g.,
video regarding vaccine handling,
storage/cold-chain management, preparation,
disposal) that are consistent with the
authorized emergency use of the vaccine as
described in the letter of authorization and
authorized labeling, without FDA’s review and
concurrence, when necessary to meet public
health needs during an emergency. Any
instructional and educational materials that
are inconsistent with the authorized labeling
are prohibited.
E. Pfizer Inc. may request changes to this
authorization, including to the authorized
Fact Sheets for the vaccine. Any request for
changes to this EUA must be submitted to
Office of Vaccines Research and Review
(OVRR)/Center for Biologics Evaluation and
Research (CBER). Such changes require
appropriate authorization prior to
implementation.14
14 The following types of revisions may be
authorized without reissuing this letter: (1)
changes to the authorized labeling; (2)
non-substantive editorial corrections to this
letter; (3) new types of authorized labeling,
including new fact sheets; (4) new
carton/container labels; (5) expiration dating
extensions; (6) changes to manufacturing
Page 9 – Pfizer Inc.
F. Pfizer Inc. will report to Vaccine Adverse
Event Reporting System (VAERS):
• Serious adverse events (irrespective of
attribution to vaccination);
• Cases of Multisystem Inflammatory Syndrome
in children and adults; and
• Cases of COVID-19 that result in
hospitalization or death, that are reported to
Pfizer Inc.
These reports should be submitted to VAERS as
soon as possible but no later than 15 calendar
days from initial receipt of the information
by Pfizer Inc.
G. Pfizer Inc. must submit to Investigational
New Drug application (IND) number 19736
periodic safety reports at monthly intervals
in accordance with a due date agreed upon with
the Office of Biostatistics and Epidemiology
(OBE)/CBER beginning after the first full
calendar month after authorization. Each
periodic safety report is required to contain
descriptive information which includes:
• A narrative summary and analysis of adverse
events submitted during the reporting
interval, including interval and cumulative
counts by age groups, special populations
(e.g., pregnant women), and adverse events of
special interest;
• A narrative summary and analysis of vaccine
administration errors, whether or not
associated with an adverse event, that were
identified since the last reporting interval;
• Newly identified safety concerns in the
interval; and
• Actions taken since the last report because
of adverse experiences (for example, changes
made to Healthcare Providers Administering
Vaccine (Vaccination Providers) Fact Sheet,
changes made to studies or studies initiated).
H. No changes will be implemented to the
description of the product, manufacturing
process, facilities, or equipment without
notification to and concurrence by FDA.
I. All manufacturing facilities will comply
with Current Good Manufacturing Practice
requirements.
J. Pfizer Inc. will submit to the EUA file
Certificates of Analysis (CoA) for each drug
product lot at least 48 hours prior to vaccine
distribution. The CoA will include the
established specifications and specific
results for each quality control test
performed on the final drug product lot.
K. Pfizer Inc. will submit to the EUA file
quarterly manufacturing reports, starting in
July 2021, that include a listing of all Drug
Substance and Drug Product lots produced after
issuance of this authorization. This report
must include lot number, manufacturing site,
date of manufacture, and lot disposition,
including those lots that
processes, including tests or other authorized
components of manufacturing; (7) new
conditions of authorization to require data
collection or study. For changes to the
authorization, including the authorized
labeling, of the type listed in (3), (6), or
(7), review and concurrence is required from
the Preparedness and Response Team
(PREP)/Office of the Center Director (OD)/CBER
and the Office of Counterterrorism and
Emerging Threats (OCET)/Office of the Chief
Scientist (OCS).
Page 10 – Pfizer Inc.
were quarantined for investigation or those
lots that were rejected. Information on the
reasons for lot quarantine or rejection must
be included in the report.
L. Pfizer Inc. and authorized distributor(s)
will maintain records regarding release of
Pfizer-BioNTech COVID‑19 Vaccine for
distribution (i.e., lot numbers, quantity,
release date).
M. Pfizer Inc. and authorized distributor(s)
will make available to FDA upon request any
records maintained in connection with this
EUA.
N. Pfizer Inc. will conduct post-authorization
observational studies to evaluate the
association between Pfizer-BioNTech COVID-19
Vaccine and a pre-specified list of adverse
events of special interest, along with deaths
and hospitalizations, and severe COVID-19. The
study population should include individuals
administered the authorized Pfizer-BioNTech
COVID-19 Vaccine under this EUA in the general
U.S. population (12 years of age and older),
populations of interest such as healthcare
workers, pregnant women, immunocompromised
individuals, subpopulations with specific
comorbidities. The studies should be conducted
in large scale databases with an active
comparator. Pfizer Inc. will provide protocols
and status update reports to the IND 19736
with agreed-upon study designs and milestone
dates.
Emergency Response Stakeholders
O. Emergency response stakeholders will
identify vaccination sites to receive
authorized Pfizer-BioNTech COVID‑19 Vaccine
and ensure its distribution and
administration, consistent with the terms of
this letter and CDC’s COVID-19 Vaccination
Program.
P. Emergency response stakeholders will ensure
that vaccination providers within their
jurisdictions are aware of this letter of
authorization, and the terms herein and any
subsequent amendments that might be made to
the letter of authorization, instruct them
about the means through which they are to
obtain and administer the vaccine under the
EUA, and ensure that the authorized labeling
[i.e., Fact Sheet for Healthcare Providers
Administering Vaccine (Vaccination Providers)
and Vaccine Information Fact Sheet for
Recipients and Caregivers] is made available
to vaccination providers through appropriate
means (e.g., e-mail, website).
Q. Emergency response stakeholders receiving
authorized Pfizer-BioNTech COVID‑19 Vaccine
will ensure that appropriate storage and cold
chain is maintained.
Vaccination Providers
R. Vaccination providers will administer the
vaccine in accordance with the authorization
and will participate and comply with the terms
and training required by CDC’s COVID-19
Vaccination Program.
Page 11 – Pfizer Inc.
S. Vaccination providers will provide the
Vaccine Information Fact Sheet for Recipients
and Caregivers to each individual receiving
vaccination and provide the necessary
information for receiving their second dose
and/or third dose.
T. Vaccination providers administering the
vaccine must report the following information
associated with the administration of the
vaccine of which they become aware to VAERS in
accordance with the Fact Sheet for Healthcare
Providers Administering Vaccine (Vaccination
Providers):
• Vaccine administration errors whether or not
associated with an adverse event
• Serious adverse events (irrespective of
attribution to vaccination)
• Cases of Multisystem Inflammatory Syndrome
in children and adults
• Cases of COVID-19 that result in
hospitalization or death
Complete and submit reports to VAERS online at
https://vaers.hhs.gov/reportevent.html. The
VAERS reports should include the words
“Pfizer-BioNTech COVID‑19 Vaccine EUA” in the
description section of the report. More
information is available at vaers.hhs.gov or
by calling 1-800-822-7967. To the extent
feasible, report to Pfizer Inc. by contacting
1-800-438-1985 or by providing a copy of the
VAERS form to Pfizer Inc.; Fax:
1-866-635-8337.
U. Vaccination providers will conduct any
follow-up requested by the U.S government,
including CDC, FDA, or other designee,
regarding adverse events to the extent
feasible given the emergency circumstances.
V. Vaccination providers will monitor and
comply with CDC and/or emergency response
stakeholder vaccine management requirements
(e.g., requirements concerning obtaining,
tracking, and handling vaccine) and with
requirements concerning reporting of vaccine
administration data to CDC.
W. Vaccination providers will ensure that any
records associated with this EUA are
maintained until notified by FDA. Such records
will be made available to CDC, and FDA for
inspection upon request.
Conditions Related to Printed Matter,
Advertising, and Promotion
X. All descriptive printed matter,
advertising, and promotional material,
relating to the use of the Pfizer-BioNTech
COVID‑19 Vaccine shall be consistent with the
authorized labeling, as well as the terms set
forth in this EUA, and meet the requirements
set forth in section 502(a) and (n) of the
FD&C Act and FDA implementing regulations.
Y. All descriptive printed matter,
advertising, and promotional material relating
to the use of the Pfizer-BioNTech COVID‑19
Vaccine clearly and conspicuously shall state
that:
Page 12 – Pfizer Inc.
• This product has not been approved or
licensed by FDA, but has been authorized for
emergency use by FDA, under an EUA to prevent
Coronavirus Disease 2019 (COVID-19) for use in
individuals 12 years of age and older; and
• The emergency use of this product is only
authorized for the duration of the declaration
that circumstances exist justifying the
authorization of emergency use of the medical
product under Section 564(b)(1) of the
FD&C Act unless the declaration is
terminated or authorization revoked sooner.
Condition Related to Export
Z. If the Pfizer-BioNTech COVID‑19 Vaccine is
exported from the United States, conditions C,
D, and O through Y do not apply, but export is
permitted only if 1) the regulatory
authorities of the country in which the
vaccine will be used are fully informed that
this vaccine is subject to an EUA and is not
approved or licensed by FDA and 2) the
intended use of the vaccine will comply in all
respects with the laws of the country in which
the product will be used. The requirement in
this letter that the authorized labeling
(i.e., Fact Sheets) be made available to
vaccination providers, recipients, and
caregivers in condition A will not apply if
the authorized labeling (i.e., Fact Sheets)
are made available to the regulatory
authorities of the country in which the
vaccine will be used.
Conditions With Respect to Use of Licensed
Product
AA. COMIRNATY (COVID-19 Vaccine, mRNA) is now
licensed for individuals 16 years of age and
older. There remains, however, a significant
amount of Pfizer-BioNTech COVID-19 vaccine
that was manufactured and labeled in
accordance with this emergency use
authorization. This authorization thus remains
in place with respect to that product for the
previously-authorized indication and uses
(i.e., for use to prevent COVID-19 in
individuals 12 years of age and older with a
two-dose regimen, and to provide a third dose
to individuals 12 years of age or older who
have undergone solid organ transplantation, or
who are diagnosed with conditions that are
considered to have an equivalent level of
immunocompromise).
BB. This authorization also covers the use of
the licensed COMIRNATY (COVID-19 Vaccine,
mRNA) product when used to provide a two-dose
regimen for individuals aged 12 through 15
years, or to provide a third dose to
individuals 12 years of age or older who have
undergone solid organ transplantation or who
are diagnosed with conditions that are
considered to have an equivalent level of
immunocompromise. Conditions A through W in
this letter apply when COMIRNATY (COVID-19
Vaccine, mRNA) is provided for the uses
described in this subsection III.BB, except
that product manufactured and labeled in
accordance with the approved BLA is deemed to
satisfy the manufacturing, labeling, and
distribution requirements of this
authorization.
IV. Duration of Authorization
Page 13 – Pfizer Inc.
This EUA will be effective until the
declaration that circumstances exist
justifying the authorization of the emergency
use of drugs and biological products during
the COVID-19 pandemic is terminated under
Section 564(b)(2) of the Act or the EUA is
revoked under Section 564(g) of the Act.
Sincerely,
--/S/--
____________________________
RADM Denise M. Hinton
Chief Scientist
Food and Drug Administration
Enclosures
|
Pfizer Inc.
Achtung: Frau Elisa Harkins
500 Arcola Road.
Collegeville, PA 19426
Liebe Frau Harkins:
Am 4. Februar 2020, gemäß § 564
(b) (1) (1 c) des fälligen Lebensmittel-,
Drogen- und Kosmetikgesetzes (das
FD&C-Gesetz), hat der Minister des
Gesundheitsdepartements (Department of Health
and Human Services - HHS) entschied, dass es
einen Notfall im Gesundheitswesen gibt, und
das ist für die "USA" und für "US"-Bürger im
Ausland auch ein Sicherheitsproblem,
miteingeschlossen der Coronavirus von 2019
(COVID-19). [1] Auf der Basis dieser
Definition erklärte der Gesundheitsminister
des HHS am 27.März 2020, dass die Bedingungen
für eine Notbewilligung erfüllt seien,
Medikamente und biologische Produkte gegen die
Covid-19-Pandemie zu verwenden, gemäß § 564
des Gesetzes (21 USC 360bbb-3), vorbehaltlich
der von diesem Abschnitt erteilten
Genehmigung. [2]
Am 11. Dezember 2020 erteilte
die Lebensmittel- und Medikamentenverwaltung
(FDA) eine Notbewilligung (EUA) für den
Notfallgebrauch des Pfizer-Biontech
Covid-19-Impfstoffs für die Prävention von
Covid-19 für Einzelpersonen ab 16 Jahre gemäss
Abschnitt 564 des Gesetzes. Die FDA hat dieses
Zulassungsschreiben erneuert am: 23. Dezember
2020, am 3 Februar 25 2021, und am 4 Mai 2021.
1 US-Abteilung für Gesundheits- und
Humanservices, Ermittlung eines Notfall- und
Erklärung zur öffentlichen Gesundheit, diesen
Umständen, die die Umstände bestehen, die
Berechtigungen gemäß § 564 Buchstabe b des
föderalen Lebensmittel-, Drogen- und
Kosmetikgesetzes, 21.s.c. § 360BB-3. 4.
Februar 2020.
2 US-Gesundheitsministerium, Erklärung über
die Existenz von Bedingungen, die einen
Beschluss gemäss Abbschnitt 564(b) gemäss dem
Lebensmittel-Medikamenten-Kosmetik-Gesetz
erlaubt: 21 US -C. § 360bbb-3, 85 FR 18250 (1.
April, 2020).
3 In der Revision vom 23. Dezember 2020
hat die FDA den Bezug zur Anzahl Dosen nach
deren Verdünnung von der Bewilligung entfernt.
Die Impfanweisungen wurden klarer gestaltet,
um Berichte ans System VAERS zu ermöglichen,
und weitere technische Korrekturen wurden
gemacht. Die FDA hat auch das Faktenblatt für
die Impfhersteller revidiert, indem klar
dargestellt wurde, dass die Anzahl Dosen pro
Impfstoff nach deren Verdünnung festgesetzt
wurde, und die Berichte ans VAERS-System
festgelegt wurden. Ausserdem wurde das
Faktenblatt für die Impfstoffhersteller und
für die Empfänger und das Pflegepersonal
revidiert, indem weitere Information mit der
Sicherheitsüberwachung hinzugefügt wurde, und
es kam auch neue Info über weitere
Covid-19-Impfstoffe hinzu.
4 Bei der Revision vom 25. Februar 2021
erlaubte die FDA eine Flexibilität bezüglich
dem monatlichen Eingabe der
Sicherheitsberichte und kontrollierte die
Anforderungen beim Berichten der Fehler von
Pfizer bei der Impfstoffverwaltung. Das
Faktenblatt für die Gesundheitskräfte der
Impfung (Impfhersteller) wurde mit einem
Update ergänzt. Lagerung und Transport muss
unter 0 Grad passieren. Dies wurde direkt auf
der CDC-Webseite veröffentlicht, wo auch die
Impfschäden aufgelistet sind. Dort ist auch
eine Umweltgift-Studie integriert, sowie
Impfschäden, die nach der Impfung auftreten.
Das Faktenblatt für die Empfänger und das
Gesundheitspersonal wurde revidiert und die
Impfschäden eingefügt, die nach der Impfung
auftreten.
Seite 2 - Pfizer Inc.
10, 2021,
5. Juni 25, 2021,6 und 12. August 2021.7
Am 23. August 2021 genehmigte
FDA der von der Biontech Manufacturing GmbH
eingereichten Biologics-Lizenzantrag
(BIONTech-Herstellung von Comirnaty
(Covid-19-Impfstoff, MRNA) für die aktive
Immunisierung, um die von SARS-COV-2
verursachten COVID-19 bei Einzelpersonen ab 16
Jahre zu verhindern.
Am 23. August 2021 kam die
Schlussfolgerung, dass dieses EUA unter
Abschnitt 564(g) [2] für den Schutz der
öffentlichen Gesundheit geeignet sei. Somit
gibt die FDA am 12. August 2021
eine Bewilligung heraus, wo alles revidiert
und klargestellt wird, dass die EUA bestehen
bleibt, für den Pfizer-BionTech-Impfstoff,
alles läuft weiter wie bisher mit der
vorherigen Bewilligung und Gebrauch, die
Bewilligung des Vomirnaty-Impfstofs
(Covid-19-Impfstoff, mRNA) unter Bewilligung
des EUA für gewisse Zwecke, die im bewilligten
BLA nicht enthalten sind. Ausserdem wurde auch
das Faktenblatt für die Impfhersteller
revidiert, was das Auslaufdatum des
bewilligten
Pfizer-BionTech-Covid-19-Impfstoffs angeht.
Ausserdem wurden Warnungen und
Vorsichtsmassnahmen klarer gestaltet bezüglich
Herzmuskelentzündung und Herzbeutelentzündung.
Das Faktenblatt für Empfänger und
Gesundheitspersonal wurde ebenfalls neu
gestaltet, mit den neuen Fkaten des
Faktenblatts für den bewilligten
Pfizer-BionTech-Covid-19-Impfstoff und die
Information über den vom FDA bewilligen
COMIRNATY-Impfstoff (Covid-19-Impfstoff,
mRNA).
Der Impfstoff von Pfizer-BionTech Covid-19
enthält eine Nukleosid-modifizierte
Messenger-RNA (modRNA), darin ist das virale
Spike (S)-Glycoprotein von SARS-CoV-2 in einer
Hülle mit Lipidnanopartikeln. Comirnaty
(Covid-19-Impfstoff, mRNA) ist die gleiche
Formel wie der Pfiz-Biontech
Covid-19-Impfstoff und kann mit dem
Pfizer-Biontech Covid-19-Impfstoff
austauschbar eingesetzt werden, um die
Covid-19-Impferfolge bereitzustellen. [8]
5. In der Revision vom 10. Mai 2021
bewilligte die FDA den Impfstoff von
Pfizer-BioinTech als Vorsorge gegen Covid-19
für Kinder zwischen 12 und 15 Jahren, so wie
vorher für Leute ab 16 Jahren. Ausserdem
revidierte die FDA das Faktenblatt für die
Impfstoffhersteller. Es wurde die Warnung vor
"Synkopen" (Ohnmacht) aufgenommen, die nach
der Injektion auftreten kann, vor allem bei
Jugendlichen. Die Spritzenabgabe sollte an
Orten stattfinden, um Ohnmacht zu verhindern."
Ausserdem wurde das Faktenblatt fürr die
Empfänger und das Gesundheitspersonal
revidiert mit der Aufforderung, die Fälle mit
Ohnmacht nach der Sprite zu melden.
6. Am 25. Juni 2021 hat die FDA
klare Fakten und Bedingungen bezüglich des
Exports des Covid-19-Impfstoffs aus den "USA"
festgelegt. Ausserdem wurde das Faktenblatt
für Impfstoffproduzenten revidiwert, indem
eine Warnung vor herzmuskelentzündung und
Herzbeutelentzündung nach der
Pfizer-BionTech-Impfung aufgenommen wurde. Das
Faktenblatt für Empfänger und
Gesundheitspersonal wurde mit der Information
ergänzt, dass Herzmuskelentzündung und
Herzbeutelentzündung nach der
Pfizer-BionTech-Impfung auftreten können.
7. Am 12. August 2021 kam eine Revision, bei
der die FDA eine dritte Dosis des
Pfizer-BionTech-Impfstoffs Covid-19
bewilligte, die mindestens in 28 Tagen nach
Beendigung der 2-Dosis-Regelung erfolgen muss,
bei Leuten abb 12 Jahren, die eine
Organtransplantation hatten, oder bbei Leuten
über 12, die eine Diagnose haben, die ein
gleichwertiges Immunlevel provoziert.
8. Der lizenzierte Impfstoff hat dieselbe
Formel wie der EUA-bewilligteImpfstoff, und
die Produkte sind austauschbar, um die
Impfserien auszuführen, ohne jegliche Einbusse
in Sachen Wirkung oder Sicherheit. Die
Produkte sind nur juristisch verschieden mit
Untrschiede, die die Sicherheit oder Wirkung
nicht beeinflussen.
Seite 3 - Pfizer Inc.
Am 11. Dezember 2020 wurde die Bewilligung für
Leute ab 16 Jahren erteilt., Die FDA
untersuchte die Sicherheits- und Wirkungsdaten
der laufenden 1/2/3-Versuchsphase mit fast
44.000 Teilnehmern mit einer Kontrollgruppe
mit Salzlösung im Verhältnis 1:1.
Beim Versuch sind auch Leute ab 12 dabei. Der
Bericht der FDA dieser Zeit berücksichtigte
die Sïcherheit und Wirkung mit allen Daten,
die für die Leute ab 16 erforderlich waren.
Der Bericht der FDA mit den Daten von 37.586
TeilnehmerInnen über 16, die durchschnittlich
2 Monate nach der Spritze begleitet wurden,
konnten keine speziellen Sicherheitsbedenken
feststellen, die die Erteilung einer
EUA-Bewilligung ausschliessen würden. Die
Analyse der FDA mit den erhältlichen
Wirkungsdaten von 36.523 TeilnehmerInnen ab 12
Jahren ohne SAS-CoV-2-Infektion 7 Tage nach
der zweiten Dosis (da waren 8 Covid-19-Fälle
in der Impfgruppe, verglichen mit 162
Covid-19-Fällen in der Plazebogruppe).
Basierend auf diesen Daten und mit den
Fabrikationsdaten hinsichtlich der
Produktequalität und Stetigkeit
schlussfolgerte die FDA, dass es vernünftig
wäre zu glauben, dass der Impfstoff von
Pfizer-BionTech Covid-19 wirksam sein könne.
Ausserdem definierte die FDA, es sei
vernünftig zu schlussfolgern, wenn man alle
wissenschaftliche Beweise berücksichtigt, dass
die bekannten und starken Vorteile des
Pfizer-BionTech-Covid-19-Impfstoffs die
bekannten und die potentiellen Risiken des
Impfstoffs wettmachen, als Prävention gegen
Covid-19 bei Leuten ab 16 Jahren.
Schlussendlich stimmte das Beratungskomitee
für Impfstoffe und verwandte, biologische
Produkte am 10. Dezember 2020 in diesem Sinne
für ein Abkommen.
Am 10. Mai 2021 kam die Bewillgiung für Kinder
zwischen 12 und 15 Jahren. Die FDA ging
nochmals die Daten bezüglich Sicherheit und
Wirkung durch, die von Versuch der 1-2-3-Phase
mit knapp 46.000 Teilnehmern stammten,
darunter auch 2260 Kinder zwischen 12 und 15
Jahren. Die Kontrollgruppe bekam Salzlösung im
Verhältnis 1:1.
FDAs Überprüfung der verfügbaren
Sicherheitsdaten von 2.260 Teilnehmern 12 bis
15 Jahre, die nach einem Median von 2 Monaten
nach Erhalt der zweiten Dosis folgten,
ermittelte keine speziellen
Sicherheitsbedenken, die die Ausgabe einer EUA
ausschließen würden. FDA-Analyse der
SARS-COV-2 50% neutralisierenden
Antikörpertiter 1 Monat nach der zweiten Dosis
von Pfizer-Biontech Covid-19-Impfstoff in
einer Teilmenge von Teilnehmern, die keinen
serologischen oder virologischen Anhaltspunkte
der vergangenen SARS-COV-2-Infektion hatten,
bestätigen die geometrische Der mittlere
Antikörpertiter in den Teilnehmern 12 bis 15
Jahre alt war dem geometrischen mittleren
Antikörpertiter in den Teilnehmern 16 bis 25
Jahre nicht unterlegen. Die Analyse der
FDA-Analyse der verfügbaren beschreibenden
Wirksamkeitsdaten von 1.983 Teilnehmern 12 bis
15 Jahre alt, ohne dass die
SARS-COV-2-Infektion vor 7 Tagen nach der
Dosis 2 bestätigen, dass der Impfstoff zu 100%
effektiv war (95% Konfidenzintervall 75.3,
100.0) Verhinderung von COVID-19, die
mindestens 7 Tage nach der zweiten Dosis (ohne
COVID-19-Fälle in der Impfstoffgruppe im
Vergleich zu 16 COVID-19 in der
Placebo-Gruppe) auftreten. Basierend auf
diesen Daten kam FDA zu dem Schluss, dass es
angemessen ist, zu glauben, dass
Pfizer-Biontech Covid-19-Impfstoff in
Einzelpersonen 12 bis 15 Jahre lang wirksam
sein kann. Darüber hinaus hat die FDA
festgestellt, dass es angemessen ist, auf der
Grundlage der Gesamtheit der
wissenschaftlichen Beweise zu schließen, da
die bekannten und potenziellen Vorteile des
Pfizer-Biontech-Covid-19-Impfstoffs die
bekannten und potenziellen Risiken des
Impfstoffs überwiegen, um die Prävention von
Covid- 19 in Einzelpersonen 12 bis 15 Jahre
alt.
Seite 4 - Pfizer Inc.
Für den 12. August 2021 Die Genehmigung einer
dritten Dosis des Pfiz-Biontech
Covid-19-Impfstoffs in Einzelpersonen in
Einzelpersonen, die 12 Jahre oder älter sind,
die eine solide Organtransplantation
unterzogen haben, oder Einzelpersonen 12 Jahre
oder älter, die mit Bedingungen diagnostiziert
werden, die mit Bedingungen diagnostiziert
werden Für ein äquivalentes Niveau der
Immunocompromise, überprüfte FDA die
Sicherheits- und Effektivitätsdaten, die in
zwei Manuskripten auf massiven
Organtransplantationsempfängern berichtet
wurden. Die erste Studie war eine einzelne
Armstudie, die in 101 Personen durchgeführt
wurde, die verschiedene massive
Organtransplantationsverfahren (Herz, Nieren,
Leber, Lunge, Pankreas) einen Median von 97 ±
8 Monaten zuvor durchlaufen hatten. Eine
dritte Dosis des Pfizer-Biontech
Covid-19-Impfstoffs wurde von etwa 2 Monaten,
nachdem sie eine zweite Dosis erhielt, auf 99
dieser Personen verabreicht. Die Höhepunkte
der gesamten SARS-COV-2-bindenden Antikörper,
die die vorgegebenen Erfolgskriterien für den
Erfolg erfüllen, trat vier Wochen nach der
dritten Dosis in 26/59 (44,0%) derjenigen auf,
die anfangs als seronegativ angesehen wurden,
und erhielten eine dritte Dosis des Pfizers
-BionTech Covid-19-Impfstoff; 67/99 (68%) der
gesamten Gruppe, die eine dritte Impfung
erhielt, wurden anschließend als
Antikörperniveaus angesehen, die eine
signifikante Antwort angeben. Bei denjenigen,
die eine dritte Impfstoffdosis erhielten, war
das nachteilige Ereignisprofil ähnlich, dass
nach der zweiten Dosis- und No-Grad-3- oder
Grad-4-Ereignisse berichtet wurden. Eine
unterstützende Sekundärstudie beschreibt eine
doppelblinde, randomisierte, randomisierte
Studie, die in 120 Individuen durchgeführt
wurde, die verschiedene massive
Organtransplantationsverfahren (Herz, Nieren,
Nieren-Pankreas, Leber, Lunge, Pankreas) ein
Median von 3,57 Jahren zuvor unterzogen hatten
(Reichweite 1.99) -6,75 Jahre). Eine dritte
Dosis eines ähnlichen MRNA-Impfstoffs (der
Moderna Covid-19-Impfstoff) wurde an 60
Personen in etwa 2 Monaten nachdem sie eine
zweite Dosis (d. H. Dosen um 0, 1 und 3
Monate) erhielt; Saline Placebo erhielt 60
Einzelpersonen oder -vergleich. Das primäre
Ergebnis war Anti-RBD-Antikörper bei 4 Monaten
mehr als 100 U / ml. Dieser Titer wurde
basierend auf NHP Challenge-Studien sowie
einer großen klinischen Kohortenstudie
ausgewählt, um darauf hinzuweisen, dass dieser
Antikörpertiter schützend war.
Sekundärergebnisse basierten auf einem Assay
von Virenneutralisierungsassay und
polyfunktionellen T-Zellantworten. Die
Basismerkmale waren zwischen den beiden
Studienarmen vergleichbar, ebenso wie der
Anti-RBD-Titer-Anti-Rbd-Titer und
Neutralisierung von Antikörpern. Die gesamten
SARS-COV-2-bindenden Antikörper von
SARS-COV-2, die eine wesentliche Reaktion
auftraten, trat vier Wochen nach der dritten
Dosis in 33/60 (55,0%) der Moderna
Covid-19-geimpften Gruppe und 10/57 (17,5%)
der Placebo-Individuen auf . In den 60er
Personen, die eine dritte Impfstoffdosis
erhielten, war das unerwünschte Ereignisprofil
ähnlich, dass nach der zweiten Dosis und NO NO
NOW-3- oder Grad-4-nachteiliger Ereignisse
berichtet wurden. Trotz der moderaten
Verbesserung in Antikörpertiter, der
Gesamtheit von Daten (dh unterstützendem
Papier von Halle et al. Demonstrierte die
Wirksamkeit des Produkts in älteren Menschen
und Personen mit Co-Briorities) unterstützt
die Schlussfolgerung, dass eine dritte Dosis
der Pfizer-Biontech Covid -19 Impfstoff kann
in dieser Bevölkerung wirksam sein, und dass
die bekannten und potenziellen Vorteile einer
dritten Dosis von Pfizer-Bionech
Covid-19-Impfstoff die bekannten und
potenziellen Risiken des Impfstoffs für
immunocompromisierte Individuen mindestens 12
Jahre alt, die erhalten haben, die erhalten
haben Zwei Dosen des
Pfiz-Biontech-Covid-19-Impfstoffs und die mit
einer soliden Organtransplantation unterzogen
wurden, oder die mit Bedingungen
diagnostiziert werden, die als äquivalentes
Grad der Immunocompromise angesehen werden.
Nach dem Schluss, dass die Kriterien für die
Ausgabe dieser Ermächtigung nach § 564 (c des
Gesetzes erfüllt sind, ermächtige ich den
Notfall von Pfizer-Biontech Covid-19-Impfstoff
für die Prävention von Covid-19, wie im
Geltungsbereich von Berechtigungsabschnitt
dieses Schreibens (Abschnitt II) und den
Bedingungen dieser Ermächtigung. Wie in
Abschnitt III.BB festgelegt, genehmige ich den
Gebrauch von Comirnaty (Covid-19-Impfstoff,
mRNA) im Rahmen dieser EUA, wenn er verwendet
wird, um ein Zwei-Dosis-Regime für
Einzelpersonen im Alter von 12 bis 15 Jahren
zu versorgen, oder
Page 5 - Pfizer Inc.
eine dritte Dosis an Individuen 12 Jahre alt
oder älter, die eine solide
Organtransplantation unterzogen haben oder mit
Bedingungen diagnostiziert werden, die als
gleichwertiges Maß an Immunocompromise
angesehen werden.
I. Kriterien für die Erteilung der Genehmigung
Ich bin zu dem Schluss gekommen, dass der
Notfall von Pfizer-Biontech Covid-19-Impfstoff
für die Prävention von COVID-19, wenn er, wie
im Rahmen der Berechtigung beschrieben, wie im
Rahmen der Berechtigung (Abschnitt II)
beschrieben, die Kriterien für die Erteilung
einer Genehmigung gemäß § 564 (c) erfüllt die
Handlung, weil:
A. SARS-COV-2 kann zu einer ernsthaften oder
lebensbedrohlichen Erkrankung oder einem
Zustand führen, einschließlich schwerer
Atemwegserkrankung, den Menschen, die von
diesem Virus infiziert sind;
B. Basierend auf der Gesamtheit
wissenschaftlicher Beweise für FDA, ist es
angemessen zu glauben, dass der Pfiz-Biontech
Covid-19-Impfstoff bei der Verhinderung von
Covid-19 wirksam sein kann, und das, wenn er
unter den in dieser Zulassung beschriebenen
Bedingungen verwendet wird, das bekannte und
potenzielle Vorteile des
Pfiz-Biontech-Covid-19-Impfstoffs, wenn es
verwendet wird, um zu verhindern, dass
Covid-19 seine bekannten und potenziellen
Risiken überwiegt; und
C. Es gibt keine angemessene, genehmigte und
zur Verfügung stehende 2-Alternative zum
Notfall von Pfizer-Biontech
Covid-19-Impfstoff, um COVID-19.10 zu
verhindern
II. Berechtigungsumfang.
Ich habe gemäß § 564 (d) (1) des Gesetzes
abgeschlossen, dass der Umfang dieser
Ermächtigung wie folgt begrenzt ist:
• Pfizer Inc. liefert Pfizer-Biontech
Covid-19-Impfstoff entweder direkt oder durch
autorisierte Händler, 11 bis zur
Notfallanteilinhaber12, wie von den
US-amerikanischen Rettungsantriebspakete
9 Obwohl Comirnaty (Covid-19-Impfstoff, mRNA)
zugelassen ist, um COVID-19 in Einzelpersonen
16 Jahre und älter zu verhindern, besteht
nicht ausreichender genehmigter Impfstoff, der
für die Vertrieb dieser Bevölkerung in seiner
Gesamtheit zum Zeitpunkt der Wiedergabe dieser
EUA in seiner Gesamtheit zur Verfügung steht .
Zusätzlich gibt es keine Produkte, die zur
Verhinderung von COVID-19 in Einzelpersonen 12
bis 15 genehmigt werden, oder die zur
Bereitstellung einer zusätzlichen Dosis der in
dieser EUA beschriebenen immunocompromisierten
Bevölkerung zugelassen sind.
10 Die Verordnung unter § 564 Buchstabe c (4)
des Gesetzes wurde keine anderen Kriterien der
Emission verschrieben.
11 "Autorisierte Händler (e)" werden von
Pfizer Inc. identifiziert oder, falls
zutreffend von einer US-Regierungseinheit, wie
z. B. der Zentren für Krankheitssteuerung und
-prävention (CDC) und / oder anderer
Beeintragung, als Entität oder Entitäten, die
darf Verteilen Sie den autorisierten
Pfizer-Biontech Covid-19-Impfstoff.
12 Für den Zweck dieses Schreibens bezieht
sich "Notfallanteilinhaber" auf ein Agentur
für die öffentliche Gesundheit und ihre
Delegierten, die rechtliche Verantwortung und
Autorität aufweisen, auf einen Vorfall zu
reagieren, der auf politische oder
geografische Grenzlinien (z. B. Stadt, Bezirk,
Tribal, territorial ist) , Staatlicher oder
föderaler oder fundamentaler) oder funktional
(z. B. Strafverfolgung oder öffentliches
Gesundheitsbereich) oder der Befugnis,
Impfstoff in einer Notlage zu verwalten, zu
liefern, zu liefern oder zu verteilen. In
einigen Fällen (z. B. abhängig von einer
staatlichen oder lokalen Jurisdiktion von
COVID-19-Impfergaberorganisation und -plänen)
können sich die Rollen und
Verantwortlichkeiten zwischen den
"Notfallanteilinhabern" und "Impfanbietern"
(z. B. ein lokaler Gesundheitsabteilung
handelt Verwalten von Covid-19-Impfstoffen;
Wenn eine Apotheke in einem handelt
Seite 6 - Pfizer Inc.
Seite 6 - Pfizer Inc.
Regierung, einschließlich der Zentren für
Krankheitssteuerung und -prävention (CDC) und
/ oder anderer Designee, zur Verwendung mit
den Bedingungen dieser EUA;
• Der PFIZER-BIONTECH COVID-19-Impfstoff, der
von dieser Ermächtigung abgedeckt wird, wird
von Impfanbietern13 verabreicht und nur zur
Verhinderung von COVID-19 in Einzelpersonen 12
und älter verwendet; und
• Pfizer-Biontech Covid-19-Impfstoff kann von
einem Impfanbieter ohne individuelles Rezept
für jeden Impfstoffempfänger verabreicht
werden.
Diese Ermächtigung deckt auch die Verwendung
des lizenzierten Comirnaty-Produkts
(COVID-19-Impfstoff-, MRNA) -Produkt ab, wenn
sie verwendet wird, um ein Zwei-Dosis-Regime
für Einzelpersonen im Alter von 12 bis 15
Jahren bereitzustellen, oder eine dritte Dosis
für Einzelpersonen 12 Jahre oder älter , die
eine solide Organtransplantation unterzogen
haben oder mit Bedingungen diagnostiziert
werden, die als gleichwertiges Maß an
Immunocompromise angesehen werden.
Produktbeschreibung
Der Pfiz-Biontech Covid-19-Impfstoff wird als
gefrorene Suspension in mehreren Dosisäuren
geliefert. Jede Fläschchen muss mit 1,8 ml
steriler 0,9% iger Natriumchlorideinspritzung,
USP, bevor Sie den Impfstoff bilden, verdünnt
werden. Der Pfiz-Biontech Covid-19-Impfstoff
enthält kein Konservierungsmittel.
Jede 0,3-ml-Dosis des
Pfizer-Biontech-Covid-19-Impfstoffs enthält 30
mcg einer Nukleosid-modifizierten
Messenger-RNA (modrna), die das virale Spike
(S) Glykoprotein von SARS-COV-2 kodiert. Jede
Dosis des Pfizer-Biontech-Covid-19-Impfstoffs
umfasst auch die folgenden Bestandteile:
Lipide (0,43 mg (4-hydroxybutyl) azandiyl) bis
(hexan-6,1-diyl) bis (2-hexyldecanoat), 0,05
mg 2 [ Polyethylenglykol) -2000] -n,
N-Ditetradecylacetamid, 0,09 mg
1,2-destaroyl-sn-glycero-3-phosphocholin und
0,2 mg Cholesterin), 0,01 mg Kaliumchlorid,
0,01 mg monobasies Kaliumphosphat, 0,36 mg
Natriumchlorid , 0,07 mg dibasisches
Natriumphosphatdihydrat und 6 mg Saccharose.
Das Verdünnungsmittel (0,9% ige
Natriumchlorid-Injektion) trägt ein
zusätzliches 2,16 mg Natriumchlorid pro Dosis
bei.
Offizielle Kapazität unter der Behörde der
Staatsgesundheitsabteilung, um
Covid-19-Impfstoffe zu verwalten). In solchen
Fällen wird erwartet, dass die
Berechtigungsbedingungen, die auf
Notfallanteilinhaber und Impfanbieter gelten,
erfüllt werden.
13 Für den Zweck dieses Schreibens bezieht
sich der "Impfanbieter" auf die Anlage,
Organisation, Organisation, Organisation oder
den Gesundheitsdienstleister, die vom
Notfallanteil (z. B. nicht ärztliche
Gesundheitspersonal, wie Krankenschwestern und
Apotheker gemäß dem Staatsgesetz unter einem
Dauerauftrag, der vom staatlichen
Gesundheitsbeauftragten ausgestellt wurde) zur
Verwaltung oder Bereitstellung von
Impfdienstleistungen gemäß den anwendbaren
NOT-Response-Stakeholders offiziellen
COVID-19-Impfungs- und Notfallreaktionsplanen
und der im CDC Covid-19-Impfprogramm
eingeschrieben ist. Wenn der Impfstoff aus den
Vereinigten Staaten exportiert wird, ist ein
"Impfanbieter" ein Anbieter, der dazu
berechtigt ist, diesen Impfstoff in
Übereinstimmung mit den Gesetzen des Landes zu
verwalten, in dem er verwaltet wird. Für den
Zweck dieses Schreibens bezieht sich der
"Healthcare-Anbieter" auch auf eine Person,
die von der US-amerikanischen Abteilung für
Gesundheits- und Human-Dienste der
US-amerikanischen Fach- und Humanservices
befugt ist (z. B. unter der Prep-Act-Erklärung
für medizinische Gegenmaßnahmen gegen
Covid-19), um den FDA-autorisierten
Covid-19-Impfstoff zu verwalten (zB
qualifizierte Apothekentechniker und staatlich
genehmigte Apothekenpraktikanten, die unter
der Aufsicht eines qualifizierten Apothekers
handeln). Siehe, z. B. HHS. Vierte Änderung
der Erklärung unter der öffentlichen
Bereitschafts- und Notfallvorsorge für
medizinische Gegenmaßnahmen gegen COVID-19 und
Neuaufentladung der Erklärung. 85 FR 79190 (9.
Dezember, 2020).
Seite 7 - Pfizer Inc.
Das Dosierungsregime ist zwei Dosen von
jeweils 0,3 ml, 3 Wochen auseinander. Eine
dritte Dosis kann mindestens 28 Tage nach der
zweiten Dosis der beiden Dosis-Regime dieses
Impfstoffs an Individuen 12 Jahre oder älter
verabreicht werden, die eine solide
Organtransplantation unterzogen haben, oder
Einzelpersonen 12 Jahre oder älter, die mit
Bedingungen diagnostiziert werden das gilt als
äquivalentes Maß an Immunocompromise.
Die Herstellung des autorisierten
Pfizer-Biontech Covid-19-Impfstoffs ist auf
diese Einrichtungen beschränkt, die in Pfizer
Antrag auf Ermächtigung identifiziert und
vereinbart wurden.
Das Pfiz-Biontech
Covid-19-Impfstoff-Vial-Etikett und
Kartonaufkleber sind eindeutig für
"Not-Nutzungsgenehmigung" gekennzeichnet. Der
Pfiz-Biontech Covid-19-Impfstoff ist
berechtigt, verteilt, gespeichert, ferner
umverteilt zu werden und von
Notfallanteilinhabern zu verabreicht, wenn sie
in der autorisierten Herstellerverpackung (dh
Fläschchen und Kartons) verpackt werden, trotz
der Tatsache, dass die Fläschchen-Etiketten
können keine Informationen enthalten, die
ansonsten unter dem FD & C-Tat
erforderlich sein würden.
Pfizer-Biontech Covid-19-Impfstoff ist für den
Notfall mit den folgenden produktspezifischen
Informationen autorisiert, die für
Impfanbieter und Empfänger zur Verfügung
gestellt werden müssen (als "autorisierte
Kennzeichnung" bezeichnet):
• Faserblatt für Gesundheitsdienstleister, die
Impfstoffe (Impfanbieter) verwalten
(Impfanbieter): Notfallgenehmigung (EUA) von
Pfizer-Biontech Covid-19-Impfstoff, um eine
Coronavirus-Erkrankung 2019 zu verhindern
(Covid-19)
• Impfstoffinformations-Informationsblatt für
Empfänger und Pflegekräfte über Comirnaty
(Covid-19-Impfstoff, MRNA) und Pfizer-Biontech
Covid-19-Impfstoff, um eine
Coronaviruserkrankung (Covid-19) zu
verhindern.
Ich habe nach § 564 (d 1 D) (2) des Gesetzes
abgeschlossen, dass es angemessen ist, dass
die bekannten und potenziellen Vorteile von
Pfizer-Biontech Covid-19-Impfstoff, wenn sie
verwendet werden, um COVID-19 zu verhindern
und in Übereinstimmung zu verhindern Mit
diesem Umfang der Genehmigung (Abschnitt II)
überwiegen seine bekannten und potenziellen
Risiken.
Ich habe gemäß § 564 (D) (3) des Gesetzes
abgeschlossen, basierend auf der Gesamtheit
wissenschaftlicher Beweise für FDA, dass es
angemessen ist, dass Pfiz-Biontech
Covid-19-Impfstoff wirksam ist, um COVID zu
verhindern 19 in Übereinstimmung mit diesem
Berechtigungskreis (Abschnitt II) gemäß § 564
Buchstabe c (2) (a) des Gesetzes.
Nach der Überprüfung der von FDA zur Verfügung
stehenden wissenschaftlichen Informationen,
einschließlich der Informationen,
einschließlich der Informationen, die die in
Abschnitt I beschriebenen Schlussfolgerungen
unterstützen, ist der Schluss, dass
Pfizer-Biontech Covid-19-Impfstoff (wie in
diesem in diesem Anwendungsberechtigungskreis
(Abschnitt II)) die Kriterien erfüllt, die
dargelegt sind in Abschnitt 564 (c) des
Gesetzes über die Sicherheit und potentielle
Wirksamkeit.
Der Notfall des
Pfizer-Biontech-COVID-19-Impfstoffs in dieser
EUA muss im Einklang stehen und darf nicht
übersteigen, die Bedingungen der Genehmigung,
einschließlich des Berechtigungsbereichs
(Abschnitt II) und den Genehmigungsbedingungen
(Abschnitt III). Vorbehaltlich der Bedingungen
dieser EUA und
Seite 8 - Pfizer Inc.
Unter den in der Bestimmung des Sekretärs der
HHS-Bestimmung von HHS gemäß Abschnitt 564
Buchstabe b (1) (c) beschriebene Umstände und
der Generalsekretär der entsprechenden
Erklärung von HHS gemäß Abschnitt 564 (B) (1)
ist Pfizer-Biontech Covid-19-Impfstoff
Ermächtigt, COVID-19 in Einzelpersonen 12
Jahre alt und älter zu verhindern, wie im
Rahmen der Autorisierung (Abschnitt II) im
Rahmen dieser EUA beschrieben, obwohl sie
bestimmte Anforderungen nicht erfüllt, die von
einem anwendbaren Bundesgesetz nicht
erforderlich sind.
III. Genehmigungsbedingungen.
Nach § 564 der Tat erstellte ich die folgenden
Bedingungen für diese Ermächtigung:
Pfizer Inc. und autorisierte Händler (n)
A. Pfizer Inc. und Authorized Händler (n)
stellen sicher, dass der autorisierte
Pfizer-Biontech-Covid-19-Impfstoff, wie von
der US-Regierung, einschließlich CDC und /
oder anderer Bekanntheit, verteilt, sowie die
autorisierte Kennzeichnung (dh
Informationsblätter ) Wird für Impfanbieter,
Empfänger und Pflegepersonen zur Verfügung
gestellt, die mit den Bedingungen dieses
Schreibens übereinstimmen.
B. Pfizer Inc. und autorisierte Händler (n)
sorgen dafür, dass angemessener Speicher- und
Kühlkette aufrechterhalten wird, bis er bis
zur Erhalt von Notfallanteile der
Rettungsstätten der Notfallverantwortlichen
aufrechterhalten wird.
C. Pfizer Inc. stellt sicher, dass die
Bestimmungen dieser EUA allen einschlägigen
Interessengruppen (z. B. Notfallanteilinhaber,
autorisierten Distributoren und Impfanbietern)
zur Verfügung gestellt werden, die an der
Vertrieb oder Erhalt von autorisierten
Pfizer-Biontech Covid-19-Impfstoffpfeilen
beteiligt sind. Pfizer Inc. liefert allen
relevanten Interessengruppen eine Kopie dieses
Ermächtigungsteils und kommuniziert
anschließende Änderungen, die auf dieses
Zulassungsschreiben und seine autorisierte
Kennzeichnung erfolgen könnten.
D. Pfizer Inc. kann Lehr- und
Bildungsmaterialien (z. B. Video hinsichtlich
des Impfhandlings, der Lagerung /
kaltkethärtlichen Verwaltung, der
Vorbereitung, der Entsorgung) entwickeln und
verbreiten, die mit dem autorisierten Notfall
des Impfstoffs in Einklang stehen, wie in der
Zulassungsschreiben beschrieben und
autorisierte Kennzeichnung, ohne
FDA-Überprüfung und Überprüfung, wenn nötig,
um den öffentlichen Gesundheitsbedarf während
eines Notfalls zu erfüllen. Anleitungen und
Bildungsmaterialien, die mit der autorisierten
Kennzeichnung inkonsistent sind, sind
verboten.
E. Pfizer Inc. kann Änderungen an dieser
Ermächtigung anfordern, einschließlich der
autorisierten Faktenblätter für den Impfstoff.
Jeder Antrag auf Änderungen an dieser EUA muss
dem Amt für Impfstoffe Forschung und
Überprüfung (OVRR) / Center für
Biologics-Bewertung und -forschung (CBER)
eingereicht werden. Diese Änderungen erfordern
eine entsprechende Genehmigung vor der
Implementierung.14
14 Die folgenden Arten von Revisionen können
autorisiert werden, ohne diesen Buchstaben neu
zu erneuern: (1) Änderungen an der
autorisierten Kennzeichnung; (2) nicht
wesentliche redaktionelle Korrekturen an
diesem Brief; (3) neue Arten der autorisierten
Kennzeichnung, einschließlich neuer
Faktenblätter; (4) neue Karton- /
Behälteraufkleber; (5)
Ablaufdating-Erweiterungen; (6) Änderungen an
der Fertigung
Seite 9 - Pfizer Inc.
F. Pfizer Inc. berichtet dem
Impfstoff-unerwünschten Ereignisberichtssystem
(VAERS):
• schwere unerwünschte Ereignisse (unabhängig
von der Attribution zur Impfung);
• Fälle von Multisystem Entzündungssyndrom bei
Kindern und Erwachsenen; und
• Fälle von Covid-19, die zum
Krankenhausaufenthalt oder zum Tod führen, der
dem Pfizer Inc. gemeldet wird.
Diese Berichte sollten so bald wie möglich an
VAERS eingereicht werden, jedoch spätestens 15
Kalendertage vom ersten Erhalt der
Informationen von Pfizer Inc.
G. Pfizer Inc. muss der Untersuchung der neuen
Medikamentenanwendung (INN) Nr. 19736
periodische Sicherheitsberichte in monatlichen
Abständen gemäß einem Fälligkeitsdatum
einreichen, das mit dem Amt für Biostatistik
und Epidemiologie (OBE) / CBER beginnen, der
nach dem ersten vollständigen Kalendermonat
beginnt Nach der Genehmigung. Jeder
periodische Sicherheitsbericht ist
erforderlich, um beschreibende Informationen
zu enthalten, die umfassen:
• Eine narrative Zusammenfassung und Analyse
von unerwünschten Ereignissen, die während des
Berichtsintervalls eingereicht wurden,
einschließlich Intervall- und kumulativen
Zählungen nach Altersgruppen, speziellen
Bevölkerungsgruppen (z. B. schwangerer Frauen)
und nachteilige Ereignisse von besonderem
Interesse;
• eine narrative Zusammenfassung und Analyse
von Impfstoffverwaltungsfehlern, unabhängig
davon, ob sie mit einem unerwünschten Ereignis
verbunden ist oder nicht, das seit dem letzten
Berichtsintervall identifiziert wurde;
• neu identifizierte Sicherheitsbedenken im
Intervall; und
• Seit dem letzten Bericht ergriffenen
Maßnahmen aufgrund von unerwünschten
Erlebnissen (z. B. Änderungen an
Gesundheitsdienstleister, die Impfstoffe
(Impfanbieter), das Faktenblatt
(Impfanbieter), Änderungen, die an Studien
oder Studien initiiert sind, verwalten).
H. Keine Änderungen werden in die Beschreibung
des Produkts, des Herstellungsverfahrens, der
Anlagen oder der Anlagen ohne Benachrichtigung
von FDA implementiert.
I. Alle Fertigungseinrichtungen werden den
aktuellen Anforderungen an den aktuellen
Produktionspraxis entsprechen.
J. Pfizer Inc. wird den EUA-Dateizertifikaten
der Analyse (COA) für jedes
Arzneimittelprodukt von mindestens 48 Stunden
vor der Impfstoffverteilung einreichen. Die
COA enthält die etablierten Spezifikationen
und spezifische Ergebnisse für jeden
Qualitätskontrolltest, der auf dem endgültigen
Arzneimittelprodukt läuft.
K. Pfizer Inc. wird den vierteljährlichen
Fertigungsberichten der EUA-Datei ableiten, ab
Juli 2021, darunter eine Auflistung aller
Arzneimittelsubstanz und Drogenprodukte, die
nach der Erteilung dieser Ermächtigung
hergestellt werden. Dieser Bericht muss
Losnummern, Fertigungsstätten,
Herstellungsdatum und Los-Disposition
enthalten, einschließlich derjenigen, die
Prozesse, einschließlich Tests oder andere
autorisierte Fertigungskomponenten; (7) Neue
Berechtigungsbedingungen für die Erforderung
der Datenerfassung oder -studie. Für
Änderungen an der Genehmigung, einschließlich
der autorisierten Kennzeichnung, des Typs, der
in (3), (6) oder (7) aufgeführt ist, wird die
Überprüfung und die Überprüfung und die
Überprüfung von der Bereitschafts- und
Reaktionsteam (Prep) / Büro des Center
Directors ( OD) / CBER und das Büro von
Counterterrorism und aufstrebenden Bedrohungen
(Ocet) / Büro des Hauptwissenschaftlers (OCS).
Seite 10 - Pfizer Inc.
wurden für die Untersuchung oder die Lots
unterteilt, die abgelehnt wurden.
Informationen zu den Gründen für Losquarantäne
oder Ablehnung müssen in den Bericht
aufgenommen werden.
L. Pfizer Inc. und autorisierte Händler werden
Aufzeichnungen in Bezug auf die Freisetzung
von Pfizer-Biontech Covid-19-Impfstoff für die
Verteilung (d. H. Losnummern, Menge,
Veröffentlichungsdatum) beibehalten.
M. Pfizer Inc. und Authorized Distributor (n)
werden der FDA auffordern, auf Anfrage alle in
Verbindung mit dieser EUA gepflegten
Datensätze.
N. Pfizer Inc. führt die Beobachtungsstudien
nach dem Genehmigungen zur Bewertung des
Verbandes zwischen Pfizer-Biontech
Covid-19-Impfstoff und einer vorgegebenen
Liste von unerwünschten Ereignissen von
Sonderinteresse zusammen mit Todesfällen und
Krankenhausaufenthalten sowie schweren
Covid-19 aus. Die Studienbevölkerung sollte
Einzelpersonen umfassen, die den autorisierten
Pfizer-Biontech Covid-19-Impfstoff in dieser
EUA in der allgemeinen US-Bevölkerung (12
Jahre alt und älter) verwaltet (12 Jahre alt),
Populationen von Interesse wie
Gesundheitswesen, Schwangerinnen,
immunocompromisierten Individuen,
Subpopulationen mit spezifischem
Komorbiditäten. Die Studien sollten in großen
Datenbanken mit einem aktiven Komparator
durchgeführt werden. PFIZER Inc. liefert
Protokolle und Status-Update-Berichte an den
IND 19736 mit vereinbarten
Studienkonstruktionen und Meilensteindaten.
Notfallverantwortliche
O.
P. Emergencontace-Stakeholder wird
sicherstellen, dass Impfanbieter in ihren
Rechtsordnungen sich dieses
Zulassungsschreiben bewusst sind, und die
Begriffe, die hierin und nachfolgende
Änderungsanträge, die auf das
Zulassungsschreiben vorgenommen werden,
anweisen, unterrichten sie über die Mittel,
durch die sie erhalten sollen und verwalten
Sie den Impfstoff unter der EUA und stellen
Sie sicher, dass die autorisierte
Kennzeichnung [dh das Informationsblatt für
Gesundheitsanbieter, das Impfstoff
(Impfanbieter) und das
Impfstoffinformationsblatt für Empfänger und
Pflegekräfte, den Impfanbietern durch
geeignete Mittel zur Verfügung gestellt hat
(z. B. E-Mail, Website).
Q
Impfanbieter
R. Impfanbieter verabreichen den Impfstoff in
Übereinstimmung mit der Ermächtigung und nimmt
teil und entsprechen den Bedingungen und
Schulungen, die von CDCs Covid-19-Impfprogramm
erforderlich sind.
Seite 11 - Pfizer Inc.
S. Impfanbieter bieten das
Impfstoffinformations-Faktenblatt für
Empfänger und Pflegekräfte an jede einzelne
Impfung und liefern die erforderlichen
Informationen zur Erhalt der zweiten Dosis und
/ oder dritten Dosis.
T. Impfanbieter, die den Impfstoff verwalten,
müssen die folgenden Informationen melden, die
mit der Verwaltung des Impfstoffs verbunden
sind, deren VAERS gemäß dem Informationsblatt
für Gesundheitsanbieter bewusst sind, die
Impfstoffverwaltung (Impfanbieter) verwalten:
• Impfstoffverwaltung Fehler, unabhängig
davon, ob mit einem unerwünschten Ereignis
verbunden ist oder nicht
• schwerwiegende unerwünschte Ereignisse
(unabhängig von der Attribution zur Impfung)
• Fälle von Multisystem Entzündungssyndrom bei
Kindern und Erwachsenen
• Fälle von Covid-19, die zum
Krankenhausaufenthalt oder zum Tod führen
Ergänzen und senden Sie Berichte an VAERS
online unter
https://vaers.hhhs.gov/reportevent.html. Die
VAERS-Berichte sollten die Wörter
"Pfizer-Biontech Covid-19-Impfstoff EUA" in
der Beschreibung des Berichts enthalten.
Weitere Informationen finden Sie unter
VAERS.HHS.GOV oder durch Anrufen von
1-800-822-7967. Soweit machbar, Bericht an
Pfizer Inc. durch Kontakt mit 1-800-438-1985
oder durch Bereitstellung einer Kopie der
VAERS-Form an Pfizer Inc.; Fax:
1-866-635-8337.
U. Impfanbieter werden alle von der
US-Regierung angeforderten Follow-up
durchführen, einschließlich CDC, FDA oder
einem anderen Designee, in Bezug auf
unerwünschte Ereignisse, soweit die
Notwendigkeit der Notlage möglich sind.
V. Impfanbieter überwachen und erfüllen den
Impfstoffverwaltungsanforderungen von CDC- und
/ oder Notfall-Antworten (z. B. Anforderungen
in Bezug auf das Erhalten, Nachverfolgung und
Umgang mit Impfstoff) und mit Anforderungen an
die Berichterstellung von
Impfstoffverwaltungsdaten an CDC.
W. Impfanbieter stellen sicher, dass die mit
dieser EUA verbundenen Datensätze bis zur
Benachrichtigung von FDA aufrechterhalten
werden. Solche Datensätze werden dem CDC und
der FDA für die Inspektion auf Anfrage zur
Verfügung gestellt.
Bedingungen in Bezug auf Drucksachen, Werbung
und Förderung
X. Alle beschreibenden Drucksachen, Werbung
und Werbematerial, in Bezug auf die Verwendung
des Pfizer-Biontech-Covid-19-Impfstoffs, ist
mit der autorisierten Kennzeichnung sowie den
in dieser EUA festgelegten Begriffe überein
und erfüllen die Anforderungen in Abschnitt
502 (A) und (N) des FD & C-Act- und
FDA-Durchführungsbestimmungen.
Y. Alle beschreibenden Drucksachen, Werbung
und Werbematerial, das sich auf die Verwendung
des Pfizer-Biontech Covid-19-Impfstoffs in
Bezug auf die Verwendung von Pfizer-Biontech
Covid-19 eindeutig und auffallend anfordert:
Seite 12 - Pfizer Inc.
• Dieses Produkt wurde von FDA nicht genehmigt
oder lizenziert, wurde jedoch für den Notfall
von FDA unter einer EUA ermächtigt, die
Coronavirus-Krankheit 2019 (Covid-19) für den
Einsatz in Einzelpersonen 12 Jahre und älter
zu verhindern; und
• Die Notfallnutzung dieses Produkts ist nur
für die Dauer der Erklärung zulässig, dass die
Umstände bestehen, die die Genehmigung der
Notwendigkeit des medizinischen Produkts gemäß
§ 564 (B) (1) des FD & C-Tatsächlich
bestimmen, es sei denn, die Erklärung ist
beendet oder die Ermächtigung widerrufen
früher.
Bedingung im Zusammenhang mit dem Export
Z. Wenn der Pfizer-Biontech Covid-19-Impfstoff
aus den Vereinigten Staaten exportiert wird,
sind die Bedingungen C, D und O bis Y nicht
anwenden, aber der Export ist nur zulässig,
wenn 1) die Regulierungsbehörden des Landes,
in dem der Impfstoff verwendet werden, sind
voll informiert, dass dieser Impfstoff einem
EUA unterliegt und von der FDA nicht genehmigt
oder lizenziert ist.) Die bestimmungsgemäße
Verwendung des Impfstoffs wird in jeder
Hinsicht mit den Gesetzen des Landes
eingehalten, in dem das Produkt verwendet
wird. Die Anforderung in diesem Buchstaben,
dass die autorisierte Kennzeichnung (dh
Faktenblätter) den Impfanbietern, Empfängern
und Betreuern in der Bedingung A zur Verfügung
gestellt wird, gelten nicht, wenn die
autorisierte Kennzeichnung (dh
Informationsblätter) den regulatorischen
Behörden zur Verfügung gestellt werden das
Land, in dem der Impfstoff verwendet wird.
Bedingungen in Bezug auf die Verwendung von
lizenziertem Produkt
Aa. Comirnaty (Covid-19-Impfstoff, mRNA) ist
jetzt für Einzelpersonen 16 Jahre lang
lizenziert. Es bleibt jedoch ein erheblicher
Betrag von Pfiz-Biontech Covid-19-Impfstoff,
der gemäß dieser Not-Nutzungsgenehmigung
hergestellt und markiert wurde. Diese
Ermächtigung bleibt somit in Bezug auf dieses
Produkt für die zuvor genehmigte Angabe und
Verwendung (dh zur Verhinderung von COVID-19
in Einzelpersonen 12 Jahren und älter mit
einem Zwei-Dosis-Regime eingesetzt und eine
dritte Dosis bereitzustellen Einzelpersonen 12
Jahre oder älter, die eine solide
Organtransplantation unterzogen haben oder mit
Bedingungen diagnostiziert werden, die als
äquivalentes Niveau der Immunocompromise
angesehen werden).
Bb. Diese Ermächtigung deckt auch die
Verwendung des lizenzierten Comirnaty-Produkts
(COVID-19-Impfstoff-, MRNA) -Produkt ab, wenn
sie verwendet wird, um ein Zwei-Dosis-Regime
für Einzelpersonen im Alter von 12 bis 15
Jahren bereitzustellen, oder eine dritte Dosis
für Einzelpersonen 12 Jahre oder älter , die
eine solide Organtransplantation unterzogen
haben oder mit Bedingungen diagnostiziert
werden, die als gleichwertiges Maß an
Immunocompromise angesehen werden. Bedingungen
A bis W in diesem Buchstaben gelten, als
Comirnaty (Covid-19-Impfstoff, mRNA) für die
in diesem Unterabschnitt III.BB beschriebenen
Anwendungen vorgesehen ist, mit der Ausnahme,
dass das Produkt gemäß dem genehmigten BLA
hergestellt und markiert ist, der angesehen
wird, um die Fertigung zu erfüllen,
Kennzeichnung und Vertriebsanforderungen
dieser Ermächtigung.
NS. Dauer der Genehmigung
Page 13 - Pfizer Inc.
Diese EUA wird wirksam sein, bis die Erklärung
bestehen, dass die Umstände, die die
Genehmigung der Genehmigung des Notfalls von
Medikamenten und biologischen Produkten,
während der COVID-19-Pandemie rechtfertigen,
unter § 564 (B) (2) des Gesetzes terminiert
ist, oder die EUA wird unter dem Abschnitt
widerrufen 564 (g) des Gesetzes.
Aufrichtig,
--/S/--
________________________________
Radm Denise M. Tinton
Chefwissenschaftler
Lebensmittel- und Drogenverwaltung
Gehäuse
|